A DeepMind alcançou resultado inédito e significativo na predição de estruturas de proteínas ao usar um novo sistema de IA. A empresa anunciou que seu novo algoritmo, AlphaFold2, resolveu desafios de enovelamento (ou dobra) de proteínas que tem sido um enorme desafio para os cientistas nos últimos 50 anos.
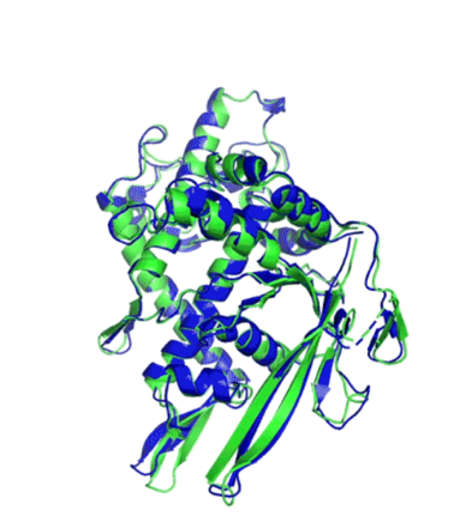
As proteínas são como blocos de construção da vida, responsáveis pela maior parte do que acontece dentro das células. Na natureza existem milhares de proteínas que fazem parte do organismo dos seres vivos, cada uma com uma forma 3D diferente que define a sua função. Identificar a estrutura da proteína é o que nos permite conhecer o que ela faz e como ela funciona. Muitos desafios do mundo estão ligados às proteínas e ao papel que desempenham, como desenvolver novos tratamentos para doenças e até mesmo encontrar alternativas para degradar lixo industrial.
No entanto, conhecemos apenas uma pequena fração da forma exata de proteínas que constituem todo o maquinário da biologia. Isso acontece porque identificar a estrutura 3D não é trivial. A proteína se forma a partir de uma sequência de aminoácidos em 1D, que vai se dobrando até formar uma estrutura complexa em três dimensões, que determina a sua função. Cada sequência de aminoácidos tem uma quantidade gigantesca de possibilidade em como se “dobrar”. É isto que dificulta predizer qual será a estrutura final da proteína.
Por muitas décadas, os cientistas vêm focando em experimentos de laboratórios para identificar estruturas de proteínas. Raio-X, cristalografia e crio-microscopia eletrônica são algumas delas, mas todas demandam um trabalho intenso no laboratório e equipamentos caros. A dificuldade é tão grande que pesquisadores passaram a buscar alternativas, como softwares de simulação e sistemas de IA para tentar ajudar a solucionar esse problema. Foi assim que surgiram duas iniciativas colaborativas super legais na Internet.
No ano 2000, alguns cientistas criaram o Folding@Home, um projeto para criar um supercomputador distribuído na Internet para trabalhar com proteínas. Na época, não havia computadores acessíveis com a capacidade de processamento que os cientistas precisavam. A ideia foi então adotar o conceito de computação voluntária, em que qualquer pessoa pudesse instalar um programa em seu computador pessoal para processar dados do projeto. A iniciativa foi considerada um sucesso. Estima-se que mais de 1 milhão de computadores ao redor do mundo fazem parte do projeto. Inclusive, a infraestrutura vem sendo utilizada para auxiliar em pesquisas da COVID-19.
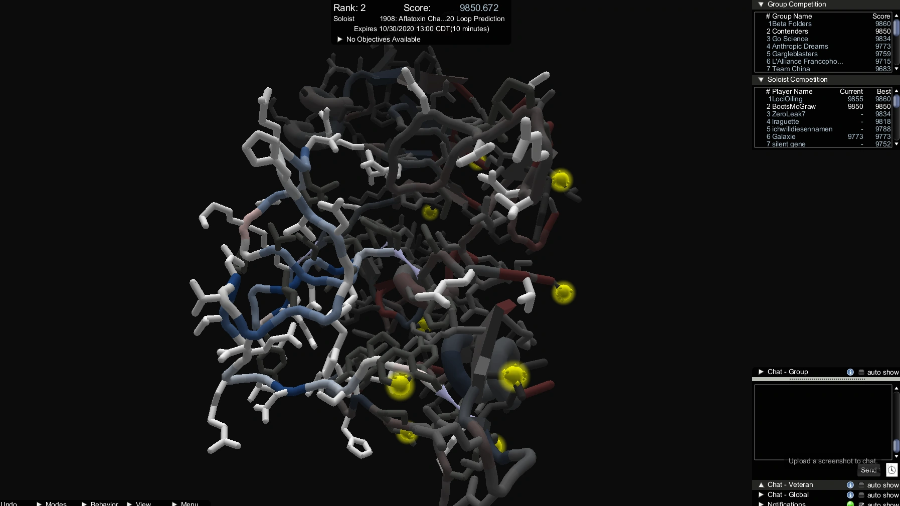
Outro projeto interessante é o Fold.it, um jogo que explora o conceito de “crowdsourcing” para que pessoas do mundo inteiro brinquem de dobrar proteínas em um ambiente 3D. Ao mesmo tempo em que se divertem, “dobrando proteínas”, as pessoas estão colaborando com a ciência a partir da descoberta de novas possibilidades de enovelamento. A iniciativa conseguiu criar uma comunidade e muitos artigos sobre o projeto foram publicados em prestigiadas revistas científicas.
Como você deve ter percebido, existem diferentes métodos e técnicas para tentar predizer a estrutura de uma proteína. Isso pode dificultar que a comunidade identifique quais são as técnicas que estão dando os melhores resultados e são mais promissoras. Pensando nisso, dois professores (John Moult e Krzysztof Fidelis) propuseram a CASP (The Critical Assessment of protein Structure Prediction), uma avaliação bienal às cegas para monitorar o progresso e estabelecer o estado da arte na previsão da estrutura de proteínas.
A CASP funciona assim: a cada dois anos, um grupo de cientistas escolhe proteínas para que outros pesquisadores ao redor do mundo façam as predições da estrutura 3D a partir da sequência de aminoácidos. Nenhum pesquisador saber de antemão qual é a estrutura correta. Eles fazem as predições utilizando suas técnicas e devolvem o resultado para que a CASP aponte os times e técnicas que tiveram o melhor resultado. Em 2018, a DeepMind alcançou o topo da tabela na competição com seu primeiro algoritmo, o AlphaFold. Foi um rebuliço na comunidade. Muitos pesquisadores não achavam que uma IA seria capaz de alcançar um resultado tão bom naquele ano.
Só que agora, em 2020, a surpresa foi ainda maior. O AlphaFold2 alcançou resultados realmente inimagináveis. O que se comenta agora é que os pesquisadores já podem utilizar as predições computacionais das estruturas como uma ferramenta padrão na pesquisa científica. É como se um novo universo tivesse sido aberto para os biólogos e químicos. O professor Venki Ramakrishan, ganhador do prêmio Nobel de química, é um dos que se mostrou empolgado com o resultado:
“Este trabalho computacional representa um avanço impressionante no problema de dobramento de proteínas, um grande desafio da biologia há 50 anos. Isso ocorreu décadas antes que muitas pessoas na área tivessem previsto. Será emocionante ver as muitas maneiras pelas quais isso mudará fundamentalmente a pesquisa biológica”
Lembra do Fold.it, o projeto em que pessoas ficavam dobrando proteínas em um jogo? Após o anúncio da DeepMind, eu acessei o fórum de discussão no site do projeto para ver o que a galera estava discutindo sobre o AlphaFold2. O que encontrei foi uma pergunta de um usuário: “A capacidade do AlphaFold de prever a dobra mudará o que estamos fazendo no fold.it? Devemos parar de dobrar proteínas?”.
Ainda não há uma resposta, mas o questionamento trouxe um sentimento nostálgico para os colaboradores. “Passamos anos dobrando proteínas e agora uma IA resolve tudo? Como vamos ficar?”, alguém pode pensar.
De fato, precisamos nos preparar para essa situação que será recorrente no futuro, já que a IA tem potencial para causar uma verdadeira revolução na ciência (e em outras áreas). Devemos dialogar mais sobre a interação entre o humano e a tecnologia, criar princípios para o uso ético e aprender a lidar com um novo “companheiro” que poderá nos ajudar em nossas atividades.
Será que um dia uma IA vai ganhar o prêmio Nobel?